In press, in the journal Vibrational Spectroscopy. In a bit of a departure from the last several terahertz (THz) papers, this study involves the simulation of the solution-phase THz spectrum of the very, very thoroughly studied 2,2'-bithiophene in solution (cis and trans geometries and lowest-frequency vibrational modes are provided in the figure below), a phase both easier and more difficult than the solid-state density functional theory (DFT) calculations that have been the mainstay of previous studies. Simplicity comes from the molecular symmetry and smaller size of the system under study, with no issues of the temperature dependence of the lattice constants or the intermolecular interaction predictions complicating the spectral assignment of the lowest frequency modes. The difficulty comes from the ability to employ multiple theoretical models to study the system and the need for far higher levels of theory in the gas phase to perform an analysis worthy of experimental comparison.
|
|
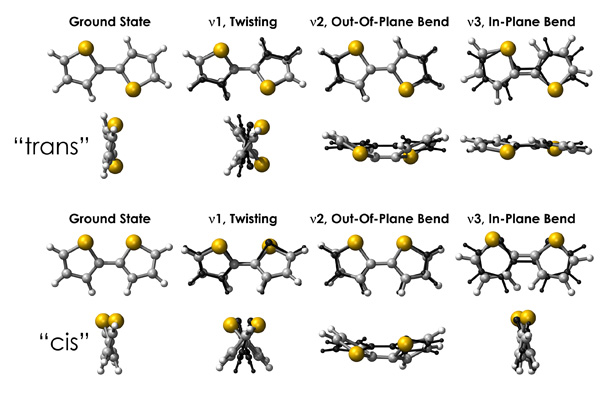
In this study, the DFT and MP2 quantum chemical calculations were used to consider molecular geometry, cis and trans conformational energy differences, rotational barrier heights, the prediction of normal mode energies, and relative peak intensities.
One topic addressed in solution that is not an issue in the crystals studied to date are the accessibility of relative conformational minima at ambient conditions (kT, room temperature). In the case of 2,2'-bithiophene, the conformational flexibility is around the exocyclic thiophene-thiophene bond. With the description of the potential energy surface (PES) for rotation about the exocyclic bond determined by conformational calculations, the second step is the determination of relative populations of the cis and trans forms as a function of temperature. In this case, weighting of the PES by the Boltzmann distribution function yields the plot shown in the bottom of the figure below, from which the relative cis and trans populations can be determined by integration of the 0 to 90 (cis) and 90 to 180 (trans) regions.
|
|
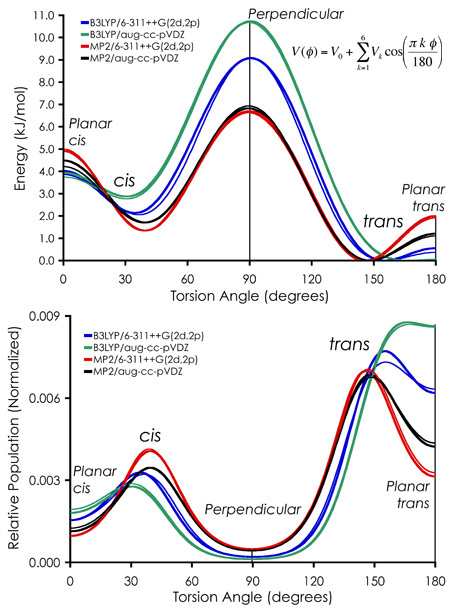
The long-short of this particular study, which I save for the article itself, is that no single theory provides all the best answers, but sufficiently high levels of theory all do settle into the reasonable vicinity of accurate. At least, to the extent that all of the experimental data is in agreement.
Anna M. Fedor1, Damian G. Allis1,2, and Timothy M. Korter1
1. Department of Chemistry, Syracuse University, Syracuse, NY 13244 USA
2. Nanorex, Inc. Bloomfield Hills, MI 48302-7188 USA
Abstract: The room temperature solution-phase terahertz (THz, 7 to 165 cm-1) spectrum of 2,2-bithiophene in cyclohexane is reported. Density functional theory (B3LYP) and ab initio (MP2) methods employing the 6-311++G(2d,2p) and aug-cc-pVDZ basis sets are used to assign the THz vibrational structure and determine the relative populations of the cis and trans conformations, as well as the trans-trans rotational barrier height and the effects of the cyclohexane solvent on the predicted molecular geometries and vibrational frequencies. Significant differences are seen in the performance of the different theoretical methods, with the best performing method dependent upon the molecular property of interest. The best fit model of the experimental THz spectrum is achieved using MP2/aug-cc-pVDZ, which places the relative trans and cis populations at 54% and 46%, respectively.
www.elsevier.com/wps/find/journaldescription.cws_home/504273/description#description
en.wikipedia.org/wiki/Terahertz
en.wikipedia.org/wiki/Thiophene
en.wikipedia.org/wiki/Density_functional_theory
en.wikipedia.org/wiki/M%C3%B8ller-Plesset_perturbation_theory
en.wikipedia.org/wiki/KT_%28energy%29
en.wikipedia.org/wiki/Boltzmann_distribution
chemistry.syr.edu
www.nanorex.com